In due articoli pubblicati giovedì su Nature e Science, la società londinese DeepMind specializzata in tecniche di deep learning e un gruppo di ricercatori guidato da David Baker, biologo strutturale della University of Washington, hanno descritto due algoritmi basati su reti neurali profonde che prevedono in modo estremamente accurato la struttura delle proteine a partire dalle sequenze di aminoacidi. In alcuni casi la loro precisione è confrontabile con quella delle strutture misurate sperimentalmente. Contestualmente hanno messo a disposizione gratuitamente il codice per il calcolo di queste strutture.
Nell'immagine: struttura cristallina del complesso proteico chaperonina. Credit: Thomas Splettstoesser/Wikimedia Commons. Licenza: CC BY-SA 3.0.
Le proteine sono le molecole fondamentali per i processi biologici e la loro struttura tridimensionale, cioè il modo in cui gli aminoacidi che le compongono sono distribuiti nello spazio, è strettamente legata alle funzioni che svolgono. Conoscere questa struttura è un compito tutt’altro che semplice. Sperimentalmente può essere estremamente oneroso e in alcuni casi impossibile. Da cinquant’anni gli scienziati cercano di sviluppare dei metodi computazionali, che inferiscano la struttura a partire dalla sequenza di aminoacidi che costituiscono la proteina. Ora la soluzione potrebbe essere a portata di mano, e non di pochi ricercatori ma della comunità intera.
In due articoli pubblicati giovedì su Nature e Science, la società londinese DeepMind specializzata in tecniche di deep learning e un gruppo di ricercatori guidato da David Baker, biologo strutturale della University of Washington, hanno descritto due algoritmi basati su reti neurali profonde che prevedono in modo estremamente accurato la struttura delle proteine a partire dalle sequenze di aminoacidi. In alcuni casi la loro precisione è confrontabile con quella delle strutture misurate sperimentalmente tramite la cristallografia a raggi X o la criomicroscopia elettronica. Contestualmente hanno messo a disposizione gratuitamente il codice per il calcolo di queste strutture, anche se con alcune differenze dal punto di vista dell’accessibilità.
Che la soluzione fosse stata sostanzialmente raggiunta era noto da dicembre del 2020, quando DeepMind aveva vinto la quattordicesima edizione della competizione Critical Assessment of techniques for protein Structure Prediction (CASP) con l’algoritmo AlphaFold2, con un grande distacco rispetto ai secondi classificati. Durante la conferenza per la presentazione dei risultati di CASP 14, John Jumper, coordinatore del gruppo che ha sviluppato AlphaFold2, aveva presentato i risultati ma senza dare troppi dettagli. Nei mesi successivi, il gruppo di David Baker ha lavorato sulle poche informazioni condivise da DeepMind per riprodurne la performance. Il risultato di questi sforzi è stato pubblicato come pre-print sulla piattaforma medRxiv all’inizio di giugno insieme all’accesso a un server su cui i ricercatori potevano testare il nuovo algoritmo, denominato RoseTTAFold, con delle sequenze di loro interesse. Lo stesso articolo è stato poi pubblicato su Science giovedì.
La precisione di RoseTTAFold è comparabile con quella di AlphaFold2, che rimane però più accurato soprattutto nella previsione delle catene laterali che in una proteina si dipartono da quella centrale, che ne costituisce l’ossatura. Sono queste catene laterali, le cosiddette side chains, a interagire con altre proteine e con altre molecole, per esempio farmaci, e dunque conoscerne con estrema precisione la struttura è cruciale per certe applicazioni. L’accuratezza di RoseTTAFold è in media di 2,5 Angstrom (un’unità di lunghezza pari a un decimo di nanometro, dell’ordine della dimensione di un atomo), mentre quella di AlphaFold2 intorno a 1,5 Angstrom, ma raggiunge anche 0,8 Angstrom per alcune strutture, di fatto eguagliando l’accuratezza sperimentale che è intorno a 0,6 Angstrom.
«Sono giornate entusiasmanti», commenta Matteo Dal Peraro, biofisico all’École Polytechnique Fédérale de Lausanne dove dirige il Laboratory for Biomolecular Modeling. «Non ci aspettavamo che DeepMind pubblicasse i dettagli dell’algoritmo e rendesse pubblico il codice così poco tempo dopo CASP 14. È il risultato della pressione che il gruppo di Baker ha esercitato presentando un proprio algoritmo ispirato ad AlphaFold2 e mettendolo a disposizione su un server». Dal momento del lancio, il server ha ricevuto 5000 richieste di previsione da parte di 500 gruppi nel mondo, spiega il giornalista Ewan Callaway su Nature.
Dal Peraro è stato uno dei valutatori della precedente edizione di CASP, quella del 2018, a cui DeepMind partecipò per la prima volta con AlphaFold, la prima versione dell’algoritmo odierno «la comunità era rimasta estremamente colpita dal loro risultato. Era la prima volta che partecipavano, e vinsero con un ottimo punteggio, già allora notevolmente superiore rispetto ai secondi classificati. Ma era solo una timida anteprima di quello che sarebbe accaduto due anni dopo. Certo, il loro atteggiamento era stato più difensivo rispetto a quello dei gruppi accademici. Avevano inizialmente titubato nel pubblicare i dettagli del loro sistema nell’edizione speciale della rivista Proteins che viene dedicata alla competizione, e avevano comunque fatto uscire un lavoro su Nature a gennaio del 2020. La pubblicazione accelerata di giovedì sembra dimostrare che l’accademia li ha convinti a giocare secondo le proprie regole».
In effetti, l’articolo di DeepMind su Nature è stato pubblicato in modalità accelerata (cioè sottoposto a peer review ma senza revisione del testo e delle figure, che restano nel formato originale), lasciando intuire che alla notizia che il lavoro del gruppo di Baker sarebbe stato pubblicato su Science, DeepMind abbia deciso di accorciare i tempi.
RoseTTAFold, anche se meno accurato, sembra però essere più veloce rispetto alla versione di AlphaFold2 presentata a dicembre scorso e richiede minori risorse computazionali. Una caratteristica fondamentale perché questo sistema diventi di uso quotidiano da parte dei ricercatori. Tuttavia, la versione del codice che DeepMind ha messo a disposizione in modalità open-source giovedì, è 16 volte più veloce di quella originale e permette di generare strutture nell’arco di alcune ore, e non giorni, ha dichiarato Jumper a Nature.
«L’esecuzione dell’algoritmo sembra essere fattibile anche su un piccolo cluster di GPU, ormai disponibile anche in università medio-piccole», conferma Dal Peraro. «Con queste caratteristiche è molto probabile che sarà in grado di rivoluzionare il lavoro quotidiano non solo dei biologi strutturali, sia computazionali che sperimentali, ma anche di biofisici e biochimici e biologi molecolari». Quando si vogliono studiare le proprietà biologiche di una proteina il primo passo è sempre la struttura, spiega Dal Peraro, e averla a disposizione quasi in tempo reale senza aspettare i tempi e sostenere i costi delle misure sperimentali, farà davvero la differenza.
L’attività onerosa dal punto di vista computazionale, ma anche di competenze, è però l’allenamento della rete neurale al cuore dei due algoritmi. «Attualmente non si ha accesso ai parametri di allenamento della rete di AlphaFold2, mentre per RoseTTAFold queste informazioni sono pubbliche», spiega Massimo Domenico Sammito, biologo computazionale che dopo una borsa Marie Curie presso il Cambridge Institute for Medical Research della University of Cambridge, lavora ora per la società biotecnologica Phoremost sempre a Cambridge. «Mi sembra comprensibile che una società privata decida di mantenere la proprietà di una parte del proprio lavoro, anche perché l’architettura e la tecnica di allenamento della rete neurale profonda sono il loro campo di specializzazione e non è escluso che in futuro possano sfruttarle in altri settori a maggior profittabilità», commenta Sammito.
AlphaFold2 ha infatti introdotto due elementi di grande novità, anche rispetto alla sua prima versione introdotta nel 2018. Il primo è il fatto che si tratta di un sistema end-to-end che minimizza cioè l’intervento degli utilizzatori. Prende in input la sequenza della proteina e di quelle a cui è legata evoluzionisticamente e restituisce come output la struttura tridimensionale. L’ottimizzazione viene effettuata sull’intera procedura, quindi partendo dalle coordinate dei singoli atomi della proteina e tornando indietro alle sequenze di input aggiustando i parametri della rete. Il secondo elemento è l’introduzione di una tecnica di apprendimento profondo sviluppata nell’ambito della visione artificiale che permette di rappresentare meglio l’interazione tra parti distanti fra loro all’interno della proteina.
RoseTTAFold, l’algoritmo sviluppato dal gruppo di Baker su ispirazione di AlphaFold2, conferma che questi elementi sono cruciali per raggiungere elevati livelli di accuratezza.
«Il campo della cristallografia a raggi X sarà forse quello che cambierà più di tutti con la diffusione su larga scala di questi algoritmi. AlphaFold2 renderà estremamente semplice l’interpretazione dei dati di diffrazione», commenta Sammito, che proprio in queste settimane sta concludendo un lavoro su questo tema. La misura di una struttura proteica tramite cristallografia a raggi X è tutt’altro che veloce e banale. Occorre infatti cristallizzare la proteina, esporla a un fascio di fotoni di alta energia disponibile solo presso gli anelli di sincrotrone (in Italia c’è ELETTRA a Trieste) e poi interpretare i dati relativi alla luce diffratta dal cristallo per tradurli in coordinate degli atomi che costituiscono la proteina.
«Resterà sicuramente più aperto il campo della criomicroscopia elettronica che permette di studiare proteine in soluzione. Interpretare le immagini raccolte con questa tecnica richiede un’elaborazione computazionale più sofisticata», commenta Sammito e spiega che ancora maggiori soddisfazioni potranno arrivare dalla tomografia crioelettronica che permetterà di studiare le proteine all’interno della cellula, seppure congelata.
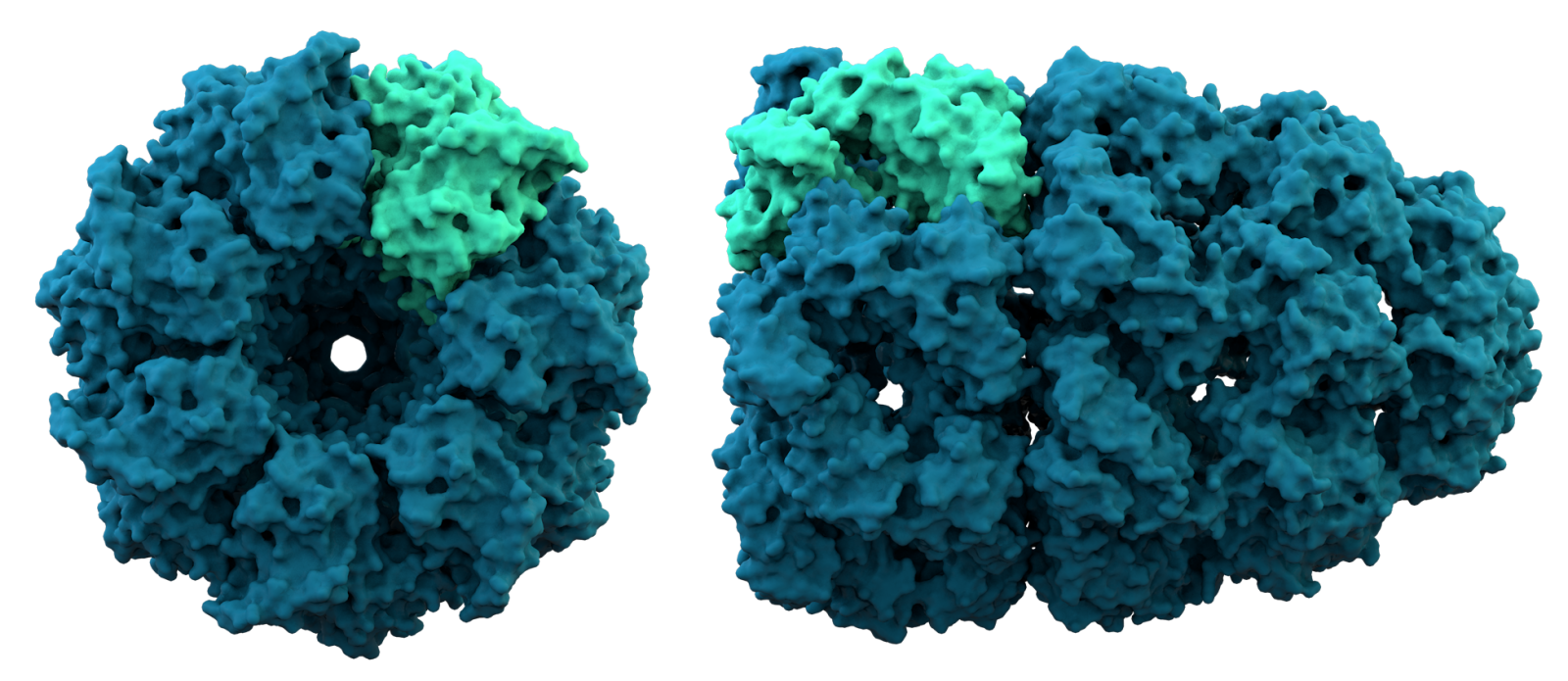
Il gruppo di Baker ha messo alla prova il suo algoritmo anche sulla previsione della struttura di complessi di proteine, cosa che AlphaFold2 non ha ancora fatto. «Prevedere la struttura dei complessi, cioè composti costituiti da più di un monomero, è estremamente importante, per esempio per capire come una proteina interagisce con altre molecole in una routine di drug discovery. Questa possibilità sarà particolarmente interessante per la ricerca in campo biomedico», commenta Sammito e aggiunge «per prevedere la struttura dei complessi, il sistema di Baker sfrutta la stessa rete neurale utilizzata per la previsione della struttura di singoli monomeri. Il fatto che questa rete funzioni bene sia per monomeri che per complessi proteici sembra suggerire che ha appreso le caratteristiche fisiche e chimiche fondamentali delle interazioni tra gli atomi all’interno di queste molecole».
Per ricevere questo contenuto in anteprima ogni settimana insieme a sei consigli di lettura iscriviti alla newsletter di Scienza in rete curata da Chiara Sabelli (ecco il link per l'iscrizione). Trovi qui il testo completo di questa settimana. Buona lettura, e buon fine settimana!

{kind=link}